Wilson's Disease
Wilson disease (WD) is an inherited copper (Cu) excretion disorder. It affects about 1 in 30,000 persons and has a carrier frequency of about 1 in 90.* Persons with WD have inherited two abnormal copies of the ATP7B gene on chromosome 13.
The ATP7B gene codes for ATPase copper transporting beta polypeptide (ATP7B). ATP7B is a cytoplasmic protein that uses ATP to transport excess Cu out of hepatocytes into the bile. Cu excreted in bile is eliminated in feces.
ATP7B also codes for the Cu binding plasma protein ceruloplasmin. Ceruloplasmin binds about 95% of plasma Cu. WD patients make lower levels of ceruloplasmin. Ceruloplasmin is also necessary for iron homeostasis. Low levels of ceruloplasmin can result in anemia.
Without functional ATP7B, toxic levels of Cu accumulate in hepatocytes. Excess Cu damages hepatocyte mitochondria causing hepatitis, accelerated apoptosis, necrosis and spillage of Cu into the blood which leads to toxic accumulation in the brain, eyes and peripheral nerves.
| Mutation of the ATP7A gene prevents Cu transport out of enterocytes resulting in severe Cu deficiency in the brain and live, known as Menkes disease. |
Adults need about 0.9 mg of Cu per day. Excess Cu is normally excreted in bile. The Western adult diet yields about 2-5 mg Cu per day.* About 40% of dietary Cu is absorbed by the GI tract the rest is excreted in feces.* Intestinal cells use ATPase copper transporting alpha polypeptide (ATP7A) to transport Cu out of the enterocyte into the portal circulation. The liver extract almost all of this Cu on the first pass. Some Cu is stored by hepatocytes and the remainder is transported by ATP7B into biliary canaliculi for excretion or bound to the plasma protein ceruloplasmin and secreted into the blood for use by the body.
The classic description of WD includes a patient at least 5 years old but less than 40 with liver symptoms, decreased serum ceruloplasmin and detectable Kayser-Fleischer rings. The actual clinical presentation of WD is highly variable. Some persons with WD have no symptoms. Most patients will experience some symptoms between the 5 and 35 years of age but they can start as early as 2 years of age or as late late as 72.*
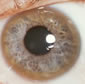 Kayser-Fleischer (K-F) rings are the result of Cu accumulation in Descemet's membrane located at the margin of the cornea. The rings are present in nearly all patients with neurological symptoms. They are absent in about 50% of patients with only liver symptoms. K-F rings are not diagnostic for WD. K-F rings are also associated with cholestatic liver disease like primary biliary cirrhosis or intrahepatic cholestasis.
Kayser-Fleischer (K-F) rings are the result of Cu accumulation in Descemet's membrane located at the margin of the cornea. The rings are present in nearly all patients with neurological symptoms. They are absent in about 50% of patients with only liver symptoms. K-F rings are not diagnostic for WD. K-F rings are also associated with cholestatic liver disease like primary biliary cirrhosis or intrahepatic cholestasis.
Forty-two percent of WD symptoms are hepatic.* Liver disease is often the initial identifying feature of Wilson disease in people between the ages of 10 and 24.* Between 20-46% of patients present with only liver symptoms. WD associated liver disease can range from asymptomatic hepatosplenomegaly with elevated transaminases to acute liver failure or WD may remain indolent until decompensated cirrhosis is apparent.*
Thirty-four percent of WD symptoms are neurological.* These symptoms often occur about 20-30 years of age with the majority of untreated patients becoming symptomatic before 50 years of age. In adulthood the most common neurologic symptoms include slurred speech (dysarthria), difficulty swallowing (dysphagia), and drooling. Other symptoms may include tremor of the head, arms, or legs; impaired muscle tone, and sustained muscle contractions that produce abnormal postures, twisting, and repetitive movements (dystonia); and slowness of movements (bradykinesia). Individuals may also experience clumsiness (ataxia) and loss of fine motor skills.
Ten percent of WD symptoms are psychiatric.* Psychiatric symptoms include: abrupt personality change, bizarre and inappropriate behavior, depression accompanied by suicidal thoughts, neurosis, or psychosis.
Ten percent of WD symptoms affect other systems including:
- Renal - aminoaciduria and nephrolithiasis
- Endocrine - hypoparathyroidism, infertility, secondary amenorrhea, repeated miscarrages
- Cardiac - arrhythmias and cardiomyopathy
- Skeletal - osteoporosis and arthritis
- Cutaneous - hyperpigmentation, lunulae ceruleae
Diagnosing WD is complicated by the number of diseases that it can mimic. Kayser-Fleischer rings can help raise suspicion of WD but they are absent in nearly half of cases with only liver symptoms. Therefore a battery of tests may be required to confirm a WD diagnosis. These tests may include:
- CBC
- Complete Metabolic Panel
- Ceruloplasmin
- Serum Cu
- 24 hour Urinary Cu
- MRI looking for white matter, cerebral and basal ganglia abnormalities
- Slit lamp examination
- Liver biopsy for Cu concentration
- ATP7B gene analysis and familial screening
Medical management of WD is focused on removing excess Cu and prevention of Cu reaccumulation. Removing excess Cu is accomplished through chelation. Chelation drugs approved for WD are D-penicillamine (Cuprimine® and Depen®) and trientine dihydrochloride (Syprine®). Chelating agents bind with certain toxic metals ions creating an inactive soluable complex that can be excreted.
D-penicillamine chelation therapy can have serious side effects. Up to 10% of patients experience autoimmune phenomena such as pemphigus, DPA-induced lupus erythematosus, polymyositis/dermatomyositis, membranous glomerulopathy and hypersensitivity pneumonitis and myasthenia. Trientine dihydrochloride is often the drug of choice for WD chelation due to its safety profile. No hypersensitivity reactions have been reported when Trientine dihydrochloride used in the normal dosage range.*
Prevention of reaccumulation is accomplished through lifelong dietary restriction and zinc therapy. Chocolate, shellfish, nuts, mushrooms and organ meat are known to be sources of Cu. Well water and water piped through copper should be evaluated for Cu content. Copper cookware and food storage containers should be avoided.*
When certain metal ions are absorbed by cells they bind to thionein creating a metallothionein. Thionein is an intracellular protein with a high affinity for physiologic metals like Cu, Zn, selenium and toxic metals like cadmium, mercury, silver, arsenic, etc. Metallothioneins are used by the cells to neutralize toxic oxidative radicals, and protect against oxidative stress. They are also used to safely store and transport physiologic metal ions within the cell for use in many physiologic processes.
Zn ions are known to stimulate production of thionein in enterocytes and hepatocytes. When Cu is absorbed by enterocytes, Cu is bound to thionein creating a metallothionein. Cu metallothioneins are stored within the enterocyte. Oral zinc acetate promotes the production of thionein. Additional intracellular thionein enables the enterocyte to safely store larger quantities of dietary Cu within the enterocyte. Stored Cu is not released into the plasma. Enterocytes have a short cell life and a rapid replacement rate. Therefore, stored Cu metallothioneins are trapped in the old cells which are shed into the intestinal lumen and excreted with feces.* Zn acetate is becoming the first line maintenance therapy due to its safety and efficacy profile.
Liver transplantation for WD is considered for patients who do not respond to medical therapy or who have acute liver failure, portal hypertension or advanced cirrhosis. Liver transplantion effectively cures Wilson's disease, with a long term survival rate of 80%.* While sucessful liver transplant can eliminate hepatic symptoms of WD the improvement of neurologic symptoms are less certain.
RnCeus
Homepage | Course
catalog | Discount
prices | Login
| Nursing
jobs | Help
©RnCeus.com