Hemophilia
Hemophilia is a bleeding disorder that occurs when a clotting factor is deficient or inactive. It is characterized by delayed blood coagulation resulting in prolonged bleeding, oozing and seepage of blood into body tissue or spaces. The severity of hemophilia is dependent upon which clotting factor is effected and the extent of the deficiency. In mild forms, the disease can go undetected until a major injury or surgical procedure reveals a coagulation defect. In severe cases, heavy bleeding occurs after minor trauma or even in the absence of injury (spontaneous bleeding). Serious complications can result from bleeding into the joints, muscles, brain, or other internal organs.
Hemophilia is most often the result of an abnormality in Factor 8 or Factor 9 (intrinsic pathway), therefore the aPTT is prolonged and is used as a screening test for the disease.
The patient with hemophilia usually has a normal PT and
a normal platelet count.
There are several types of hemophilia
 Hemophilia A, known as
classic hemophilia
Hemophilia A, known as
classic hemophilia
- Occurs about 1/5000 males worldwide
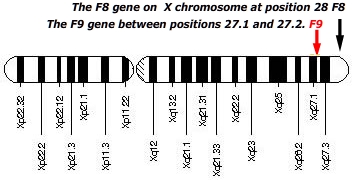
- Results from a mutation in the Factor 8 (F8) gene on the X-chromosome. The F8 gene is responsible for transcribing the coagulation factor F8. Mutations in F8 can affect the quantity and quality of factor F8 protein produced by an individual.
- More than 1,300 alterations in the F8 gene have been identified.
- Coagulation F8 is made chiefly in the liver. This protein circulates in the bloodstream in an inactive form, bound to another molecule called von Willebrand factor. In response to injury, coagulation F8 is activated and separates from von Willebrand factor. The active protein (sometimes written as coagulation VIIIa) interacts with factor IX. This interaction sets off a chain of additional chemical reactions that form a blood clot
- Diagnosis is based upon low F8 activity in the presence of normal von Willebrand factor
- Classification of hemophilia A based on in vitro clotting activity:
- Severe hemophilia A. <1% F8
- Moderate hemophilia A. 1%-5% F8
- Mild hemophilia A. 6%-35% F8
- Molecular genetic testing of F8 gene, identifies disease-causing mutations in as many as 98% of individuals with hemophilia A.
- Hemophilia B, or Christmas
disease
- Occurs in about 1/20,000 males worldwide.
- Results from a mutation in the Factor 9 (F9) gene on the X-chromosome. F9 is responsible for producing the coagulation factor IX.
- Mutations in F9 can affect the quantity and quality of factor IX protein produced by an individual.
- Bleeding episodes may be more frequent in childhood and adolescence than in adulthood.
- Hemophilia B Leyden is a variant form of hemophilia B that is characterized by postpubertal increased transcription of F9 associated with increased androgen synthesis. F9 levels of <5% of normal at birth increase to >30% by early adulthood.•
- Acquired hemophilia is a rare and severe bleeding disorder that unexpectedly occurs in about 1 per million people per year. It is caused by antibodies which inhibit the action of clotting factors, usualy F8 or F9. In about 50% of cases the cause of antibody synthesis remains unknown. The remaining 50% of cases have been attributed to conditions including: pregnancy, autoimmune or collagen vascular disorders, malignancy, drugs, respiratory disorders and infections. Treatment of acquired hemophilia focuses on minimizing complications and removal of the inhibitor.•
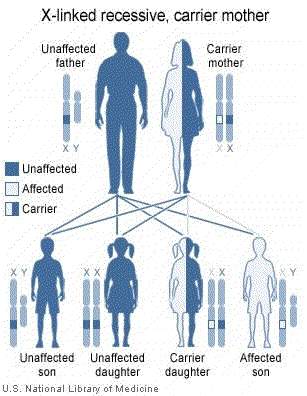
 Hemophilia A and hemophilia B can result from spontaneous or inherited X chromosome mutations. Inherited hemophilia A and B are passed in an X-linked recessive pattern. In males, a significant mutation in the one copy of the X-linked gene responsible for transcribing F8 of F9 is sufficient to cause the condition. Females have two copies of the F8 and F9 genes, therefore both copies would have to be deficient to cause the disorder. Because it is unlikely that females will have two deficient copies of this gene, it is very rare for females to have hemophilia.
Hemophilia A and hemophilia B can result from spontaneous or inherited X chromosome mutations. Inherited hemophilia A and B are passed in an X-linked recessive pattern. In males, a significant mutation in the one copy of the X-linked gene responsible for transcribing F8 of F9 is sufficient to cause the condition. Females have two copies of the F8 and F9 genes, therefore both copies would have to be deficient to cause the disorder. Because it is unlikely that females will have two deficient copies of this gene, it is very rare for females to have hemophilia.
A characteristic of X-linked inheritance is that fathers do not pass X-linked traits to their sons. In X-linked recessive inheritance, a female with one altered copy of the gene in each cell is called a carrier. Carrier females have about half the usual amount of coagulation F8 or coagulation factor IX, which is generally enough for normal blood clotting. However, about 10 percent of carrier females have less than half the normal amount of one of these coagulation factors; these individuals are at risk for abnormal bleeding, particularly after an injury, surgery, or tooth extraction.•
Treatment
Hemophilia A can be cured by a liver transplant, but the risks involved in the surgery and the requirement for lifelong medications to prevent rejection of the new liver usually outweigh the benefits for all those without liver disease. For those who are not liver transplant candidates, anti-hemophilic factor replacement products are the treatment of choice. In general, administration of anti-hemophilic factor is indicated for hemophilia when a bleeding episode arises (demand treatment) or when bleeding is anticipated or likely (prophylactic treatment).
The goal of prophylactic therapy is to intervene early and to maintain F8 or F9 above the 1% level between doses. In cases with repeated episodes of spontaneous bleeding, continuous prophylactic therapy may be needed.
A small number of patients with a normal F8 or F9 genes and about 20% of hemophilia A patients develop inhibitor antibodies to F8 or F9. Currently the management of clotting factor inhibition takes two forms:
RnCeus
Homepage | Course
catalog | Discount
prices | Login
| Nursing
jobs | Help