 Nociceptive pain can be characterized as:
Nociceptive pain can be characterized as:
- Painful stimulus that causes an organism to adopt protective behaviors that promote healing.
- Pain analogous to an alarm that signals actual or potential tissue damage, e.g., postoperative pain.
- Pain with a well defined onset associated with tissue injury from surgery, trauma, or disease related injury including inflammation.
- Pain proportional to trauma which decreases as healing occurs.
- Somatic nociceptive pain is often described as well-localized sharp, crushing, tearing pain that usually follows a dermatomal pattern.
- Visceral nociceptive pain is poorly localized dull, cramping, or colicky pain associated with peritoneal irritation, dilation of smooth muscle or a tubular passage.
- Referred visceral pain radiates in a somatic dermatomal pattern •
Nociceptive pain occurs in 5 phases: 1) Transduction, 2) Conduction, 3) Transmission, 4) Modulation, 5) Perception.
Instant Feedback:
- Transduction begins when peripheral terminals of nociceptive C fibers and A-delta (Aδ) fibers are depolarized by noxious mechanical, thermal, or chemical energy. The membranes of these terminals contain proteins and voltage-gated ion channels that convert thermal, mechanical, or chemical energy into an action potential (AP). Nociceptor terminals are spread densely throughout the skin. They are found less on periosteum, joints, tendons, muscles, and least on the surface of organs.
Normally, nociceptor terminals have a high activation threshold. They requiring intense stimulation to generate an AP. For example, thermal nociceptors are only activated by temperature extremes (>45°C or < 5°C). However, nociceptors can be made more sensitive to stimuli. Injury to neurons and surrounding tissues expose neighboring nociceptors to irritating substances, including: neurotransmitters, ATP, prostanoids, bradykinin, serotonin, histamine, and hydrogen ions (acid pH), etc. These substances lower the nociceptor's activation threshold (sensitize), creating a condition of hyperesthesia.
Hyperesthesia is a term that encompasses both allodynia and hyperalgesia. A common example of allodynia is the painful response to touch in an area of a 1st degree burn, e.g. sunburn. Normally, allodynia subsides as healing progresses. Hyperalgesia, on the other hand, results from prolonged hyperstimulation which can cause structural and functional changes to both peripheral and central neurons. These changes can cause central sensitization leading to the development neuropathic pain.
Conduction of an AP is the second phase of nociception. An AP generated in nociceptor terminals is conducted across the peripheral process to the central process were it depolarizes the presynaptic terminal. The presynaptic terminal interfaces with a network of interneurons and second order neurons in the dorsal horn. Interneurons can facilitate or inhibit transmission to second order neurons.•
There are 2 types of nociceptor fibers that conduct APs to the spinal cord. A-delta fibers (Að) are slow, thin, myelinated fibers associated with sharp/pricking, well localized pain. C fibers are very slow, thin, unmyelinated fibers that are associated with a dull, aching, throbbing, diffuse pain. A third type of fiber, A-beta (Aß), is a fast, large diameter, myelinated fiber that carries APs from mechanoreceptors. Aß fibers are believed to modulate C and Að activity within the dorsal horn.
The dorsal horn is divided into distinct gray matter laminae. Primary afferent presynaptic terminals project into specific laminae.
- Lamina I receives presynaptic terminals of Að and C fibers. It also contains second order neurons that project to the thalamus and relay to somatosensory cortex and cingulate cortex.
- Lamina II receives presynaptic terminals from C fibers which synapse on interneurons, many of which supply inhibitory signals
to lamina I projection neurons.
- Lamina III receives presynaptic terminals from Aβ fibers and dendrites from lamina V projecting neurons.
- Lamina IV receives presynaptic terminals from Aβ fibers and dendrites from lamina V projecting neurons.
- Lamina V receives presynaptic terminals of Að and contains second order neurons that project to the locus coeruleus, parabrachial nucleus, amygdala and hypothalamus.
Nociceptors serving the head and orofacial areas take a different path than from those of the rest of body. These nociceptors projects from the trigeminal ganglion and enter the CNS at the level of the pons. Trigeminal Að and C fibers pass down through the pons into the medulla where they synapse on second order neurons which then rise to decussate within the pons and pass to the thalamus.
- Transmission, the third phase begins when a nociceptive AP reaches the presynaptic terminal in the dorsal horn. The AP causes the presynaptic terminals of Að and C fibres to release a variety of pro-nociceptive substances into the synaptic cleft. C-fiber presynaptic terminals are known to release glutamate which activates postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors; substance P (SP), which activates postsynaptic NK1 receptors; and calcitonin gene–related peptide (CGRP), which activates postsynaptic CGRP receptors.•
Activation of postsynaptic receptors results in an influx of ions that depolarize second order neurons and interneurons. When a secondary neuron is depolarized it generates an action potential that is relayed through the contralateral spinothalamic tract (STT) or to the medulla and brain stem via the spinoreticular (SRT) & spinomesencephalic (SMT) tracts or to the hypothalamus via the spinohypothalamic tract (SHT).•
Postsynaptic terminals of second order neurons exhibit an array of neurotransmitter receptors. An unusual and important receptor is the N-methyl-D-aspartate (NMDA)-linked channels. NMDA channels are normally inactive because they are ordinally blocked by magnesium ions. However, intense or prolonged periods of depolarization can release the magnesium ion from the NMDA-linked channel. Loss of the Mg blockade allows an influx of calcium ions. Increased intracellular calcium ions has two important effects on the development of neuropathic disease:
- The first effect leads to a lowered activation threshold, a change in cation channel kinetics, and the insertion of more receptors in the postsynaptic membrane.
- The second effect initiates a cascade that results in the production and release of nitric oxide into the synaptic cleft. Presence of nitric oxide in the synaptic cleft causes an exaggerated release of neurotransmitters from the presynaptic terminal resulting in synaptic hyperexcitability.
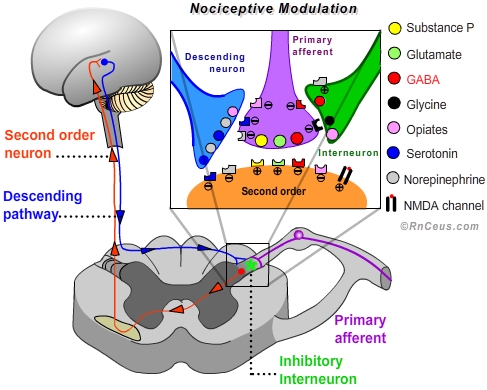
- Modulation of nociceptive transmission is an adaptive process involving both excitory and inhibitory mechanisms. "In the normally functioning state, the responses of second order neurons can be suppressed or facilitated dependent on other events important to the organism."•
For our purposes we will focus on the processes that inhibit or suppress transduction, conduction or transmission, thereby interrupting or diminishing the perception of pain.
- Peripheral modulation can be accomplished by:
a) inhibiting the sensitization of nociceptor terminals with medications such as cyclooxygenase inhibitors, e.g. aspirin, ibuprofen, etc.
b) inhibiting depolarization and repolarization of the axonal membrane. Local anesthetics like lidocaine prevent the generation or conduction of an action potential by blocking the influx of sodium through voltage-gated sodium channels located along first and second order afferents.
c) inhibiting the inflammatory response to trauma with hydrocortisone. Hydrocortisone is believed to stimulate the production of lipocortin-1, which inhibits the biosynthesis of prostaglandins and leukotrienes from arachidonic acid by inhibiting the cytosolic enzyme phospholipase A2. Lipocortin-1 is also believed to inhibit leukocytic inflammatory events including: epithelial adhesion, emigration, chemotaxis, phagocytosis.•
d) stimulating the large fast Aβ fibers in the area of injury can induce interneurons in the dorsal horn to release GABA and glycine which inhibit the release of glutamate from the primary afferent terminal, thereby preventing depolarization of the second order neuron. Mechanical stimulation and transcutaneous electrical nerve stimulation (TENS) are believed to reduce the perception of pain by activating fast Aβ fibers- Central modulation of nociception involves multiple sites and mechanisms:
a) Exogenous opioids like hydromorphone, morphine and oxycodone produce analgesia by mimicking endogenous endorphins. Opioids are able to activate the endorphin receptors: Mu, Kappa and Delta. Mu receptors are responsible for most of the analgesic effect of opioids and are present on neurons in the spinal cord, brainstem and midbrain.
• In the dorsal horn, Mu receptor act by closing voltage sensitive calcium channels on the primary afferent presynaptic terminal. Blocking the influx of Ca++ inhibits depolarization and the subsequent release of the neurotransmitters glutamate and substance P. Mu receptors also increase the efflux of K+ from the second order postsynaptic terminal, which increases the internal (-) charge, creating a hyperpolarized state.
• In the brainstem and midbrain, activated Mu receptors turn off GABAergic interneurons responsible for suppressing the antinociceptive decending pathway. In other words, opioids enhance the activity of the descending pathway which results in increased release of antinociceptive serotonin and norepinephrine from the descending neuron terminals into the dorsal horn.•
b) Endogenous opioids (endorphins) work in a manner similar to exogenous opioids. The existence of endorphins has been demonstrated by the application of stimulation produces analgesia (SPA). Electrical stimulation of the periaqueductal gray (PAG) elicits the release of endorphins which produce an analgesia that can be blocked by the opioid antagonist naloxone. When opioids are injected into the PAG, analgesia is produced via the descending pathway.•
c) Antidepressants are believed to enhance the analgesic activity of the descending pathway by increasing the availablity of synaptic monoamines. The monoamines serotonin and norepinephrine are the primary neurotransmitters released by descending pathway neuron terminals. Descending pathway neurons arise in the brainstem and terminate in close proximity to primary afferent terminals, interneurons and synaptic membrane of second order neurons located in the dorsal horn.
• Stimulation of the nucleus raphe magnus in the brainstem results in antinociception attributed to the release of serotonin (5-HT) within the dorsal horn. "Agents that block 5-HT synthesis attenuate stimulation-produced analgesia, and the application of some 5-HT agonists in the spinal cord results in inhibition of cells responsive to nociceptive stimuli." Selective serotonin-reuptake inhibitors can contribute to the modulation of nociception.•
• Stimulation of the locus coeruleus in the medulla results in antinociception attributed to the release of norepinephrine within the dorsal horn. Presynaptically noradrenaline increases inhibitory transmitters from interneurons and depresses glutamate release from both Aδ and C afferent terminals.•
- Perception of nociceptive pain is dependant upon neural processing in the spinal cord and several brain regions. Pain becomes more than a pattern of nociceptive action potentials when they reach the brain. Action potentials ascending the spinothalamic tract are decoded by the thalamus, sensorimotor cortex, insular cortex and the anterior cingulate to be perceived as an unpleasant sensation that can be localized to a specific region of the body. Action potentials ascending the spinobulbar tract are decoded by the amygdala and hypothalamus to generate a sense of urgency and intensity. It is the intergration of sensations, emotions and cognition that result in our perception of pain.
Positron Emission Tomography (PET) and Functional Magnetic Resonance Imaging (fMRI) have enable researchers to monitor perfusion, metabolism and the sequence of activity across multiple brain structures in response to painful stimulation. These tests demonstrate significant variation in the pattern, intensity and volume of brain activity between individuals exposed to similar painful stimulation. The findings reinforce the notion that pain is a complex individual experience that can not be quantified by anyone other than the person experiencing it.